Last week I talked to Vagheesh Narasimhan about his work on autozygosity in consanguineous pedigrees (Narashimhan et al. 2015), and whether you could identify pedigrees from the distribution of autozygous chunks in a single individual. That reminded me of the Altai Neanderthal paper (Prufer et al 2014), which concluded, based on the length of homozygous chunks in the genome, that the Neanderthal probably had an inbreeding coefficient of 0.125, but that you couldn’t distinguish between the possibilities leading to that: I.e. mating between half-sibs, uncle-niece, aunt-nephew, grandfather-granddaughter, grandmother-grandson or double first cousins (…the Aristocrats!).
HBD chunk distributions
The total amount of the genome (12.5%) expected to be autozygous (i.e. homozygous-by-descent; HBD) in these six cases is the same (12.5%), but in theory you could distinguish between them because the distribution of chunk lengths would be different. To look at this, I ran some simulations (code). Here’s an example (of double cross cousins). On the left is the pedigree, and on the right is a simulated genome with chunks coloured according to the founder they were inherited from:
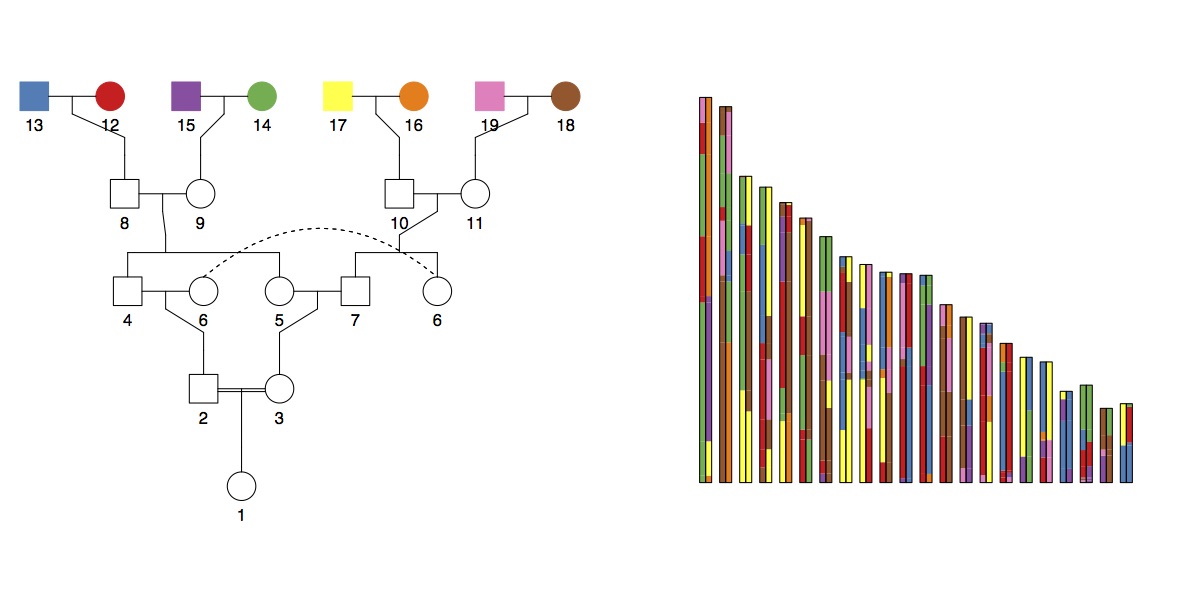 Then, you can easily extract the HBD segments and estimate the
distribution. The distribution of segment lengths in the four cases
(excluding two that are just sex-reversed) look like this:
Then, you can easily extract the HBD segments and estimate the
distribution. The distribution of segment lengths in the four cases
(excluding two that are just sex-reversed) look like this:
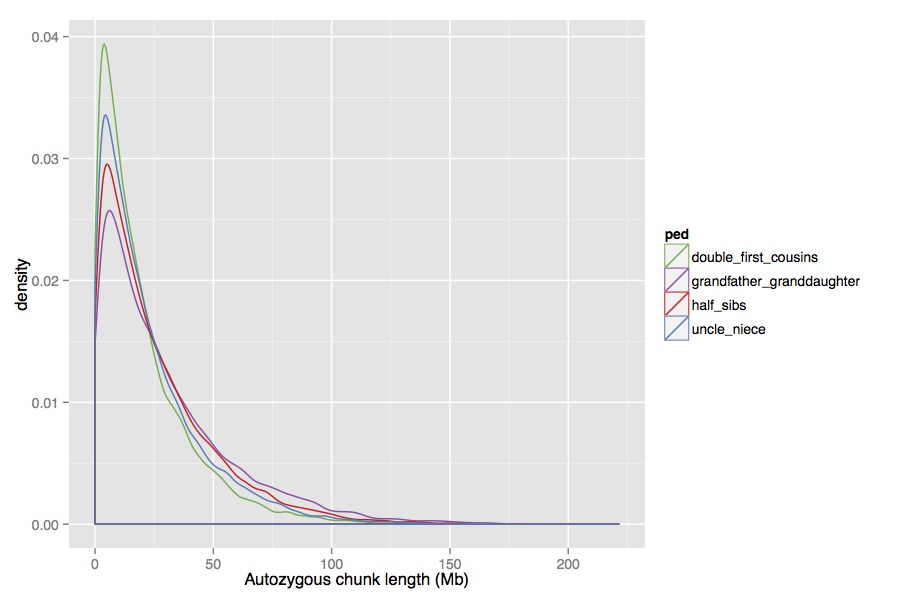
There is a difference, but it’s quite subtle. One thing to note is that I used sex-specific recombination rates (from Fig.5 of Fledel-Alon et al 2011), which lets you distinguish between, say, half-sibs, and grandfather-granddaughter, which you otherwise wouldn’t be able to. Overall though, these distribution are very similar and it seems like it would be pretty hard to tell the difference between them, given that you don’t get many chunks from a single individual.
I simulated under each model and tested whether you could ever reject any of the other models using a Kolmogorov-Smirnoff test, and the answer was basically no. This seems consistent with Supplementary Figure 10.2 of Prufer et al, which seems to say that the models can’t be reliably distinguished.
It seems like you can do a lot better at reconstructing the pedigree if you have the distribution of IBD chunks over a small number of individuals though (Staples et al 2011).
References
Narasimhan et al. 2016. Health and population effects of rare gene knockouts in adult humans with related parents.
Prufer et al 2014. The complete genome sequence of a Neanderthal from the Altai Mountains.
Fledel-Alon et al 2011. Variation in human recombination rates and its genetic determinants.
Staples et al 2011. PRIMUS: Rapid Reconstruction of Pedigrees from Genome-wide Estimates of Identity by Descent